细胞自噬是真核细胞中保守的降解性代谢通路,对维持细胞内稳态及生存起着至关重要的作用,自噬的异常与诸多疾病的发生发展密切相关,例如癌症等。多年来,领域内的科学家一直在试图靶向自噬来治疗癌症。但目前尚未有特异性靶向自噬而治疗癌症的药物被成功开发,其重要原因是导致自噬失调的具体分子事件,特别是癌症相关自噬和生理性自噬(如饥饿诱导的自噬)之间的差异尚未阐明。随着对自噬在肿瘤发生发展过程中调控作用的研究,靶向自噬的肿瘤治疗逐渐变得重要,现在已经报道了通用靶向溶酶体(自噬)的药物氯喹作为肿瘤治疗的自噬通用药物,但是氯喹的非特异性会影响到很多生理过程,例如会抑制机体基础自噬和机体的免疫系统,导致靶向自噬抑制肿瘤的临床应用遇到瓶颈。
3月7日,清华大学生命科学学院葛亮课题组、药学院张敏课题组与广州医科大学冯杜课题组合作,以“致癌性RAS通过P38-ULK1-PI4KB轴诱导一种独特形式的非经典自噬”(Oncogenic RAS Induces a Distinctive Form of Non-Canonical Autophagy Mediated by the P38-ULK1-PI4KB Axis)为题在《细胞研究》(Cell Research)上发文。研究报道了RAS突变肿瘤过程中异常自噬的调控机制(图1),并发现异常自噬的特有细胞器,揭示了靶向PI4KB磷酸化可以特异性抑制RAS突变肿瘤的发生发展。与传统自噬通用药物氯喹作用不同,Tat-Pep.1促进CD8+T细胞的浸润。
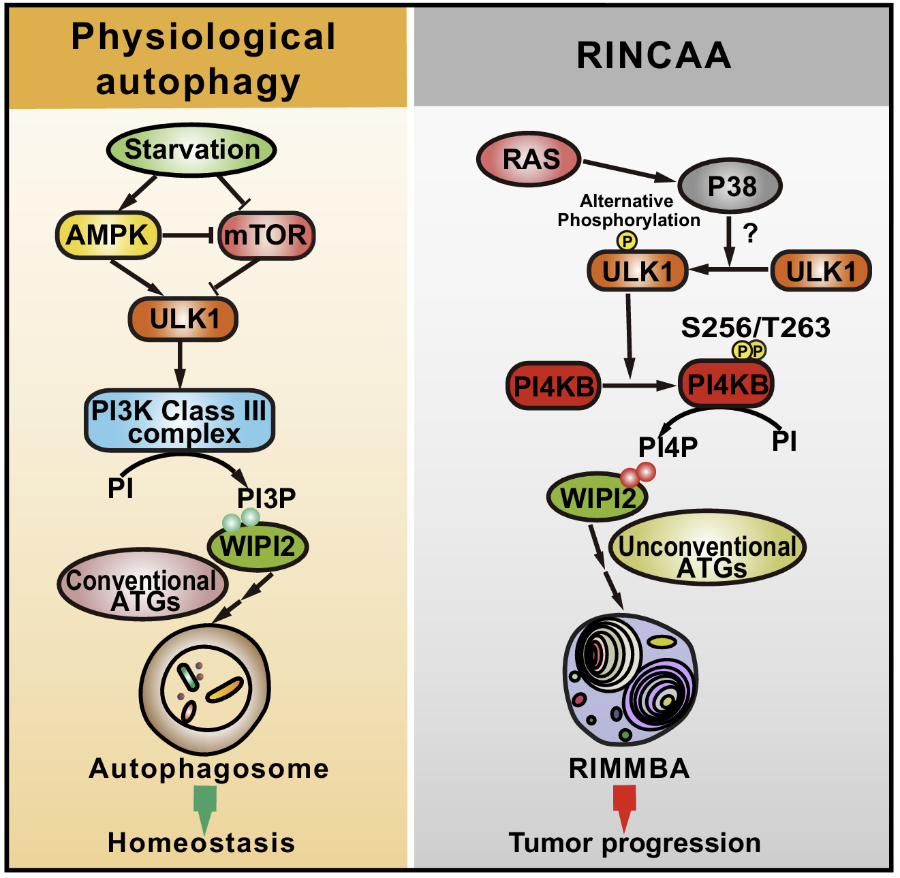
图1.RINCAA调控机制
研究人员以RAS突变的癌症模型探索自噬在肿瘤中的详细分子机制,发现了一种由RAS突变表达诱导的非经典自噬,命名为RINCAA (RAS-Induced non-canonical autophagy via ATG8ylation)。与饥饿诱导的自噬相比,RINCAA受不同的自噬因子调控,形成ATG8家族蛋白(如LC3和GABARAP)标记的多膜泡和多层结构的非经典自噬体结构,将其命名为RIMMBA(RAS-induced multivesicular/multilaminar bodies of ATG8ylation)(图2)。
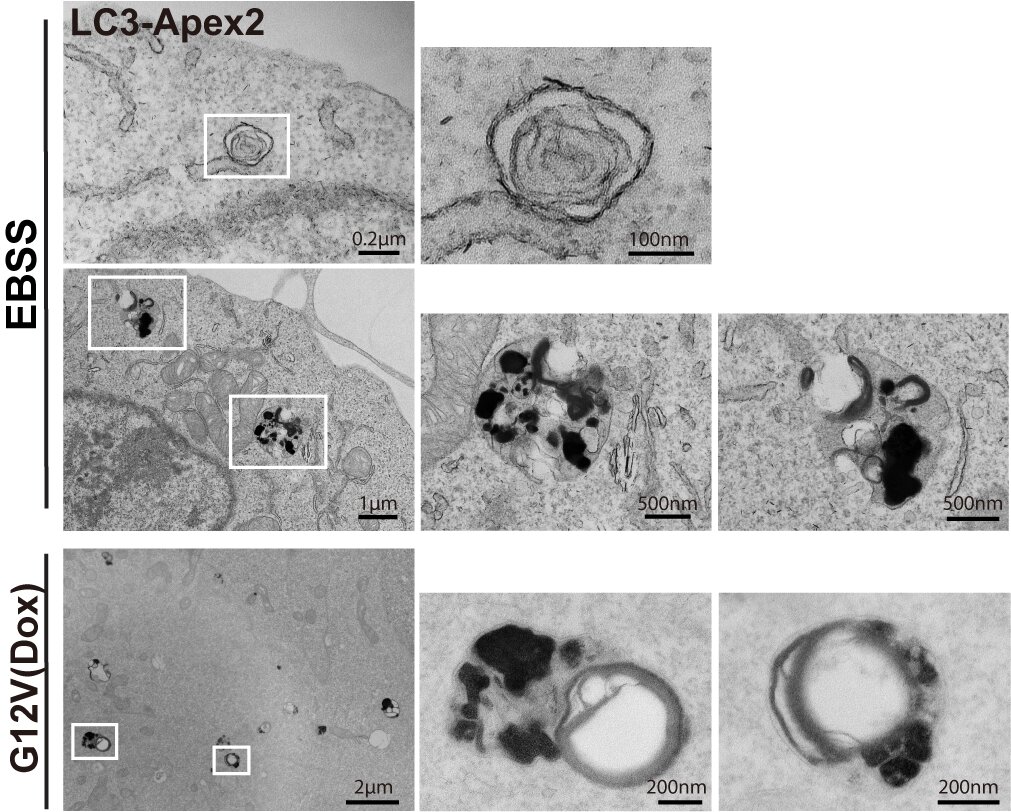
图2. 电镜观察饥饿诱导的自噬体与RAS诱导的RIMMBA RINCAA的一个显著特征是经典自噬通路中的PI3K被替换为PI4KB。研究者鉴定了P38-ULK1-PI4KB-WIPI2级联信号控制这一过程,KRAS激活下游P38通路,进一步促进ULK1的激活。激活的ULK1促进PI4KB在S256和T263位点磷酸化并促进PI4P的产生,WIPI2作为PI4P效应器招募ATG16L1/ATG12-ATG5酯化复合体促进ATG8ylation和非经典自噬体的形成
在RAS突变的肿瘤细胞系与结直肠癌病人中,PI4KB在S256与T263位点的磷酸化水平均显著上调,表明PI4KB在S256与T263位点的磷酸化可能作为RAS突变肿瘤治疗的一个非经典自噬的特异性靶点。研究人员设计并合成的Tat-Pep1底物肽可以靶向抑制PI4KB在S256与T263位点的磷酸化进而抑制RINCAA活性,最终抑制KPC胰腺癌模型的肿瘤生长,延长KPC胰腺癌模型生存期,效果比自噬通用药物氯喹更好(图3)。Tat-Pep1底物肽靶向RINCAA的高特异性同时保持机体的免疫活性,奠定了PI4KB磷酸化可作为一种非常有潜力的靶向自噬的肿瘤治疗靶点。未来基于靶向PI4KB在S256和T263磷酸化的小分子药物也将为临床RAS突变肿瘤病人带来新的希望。
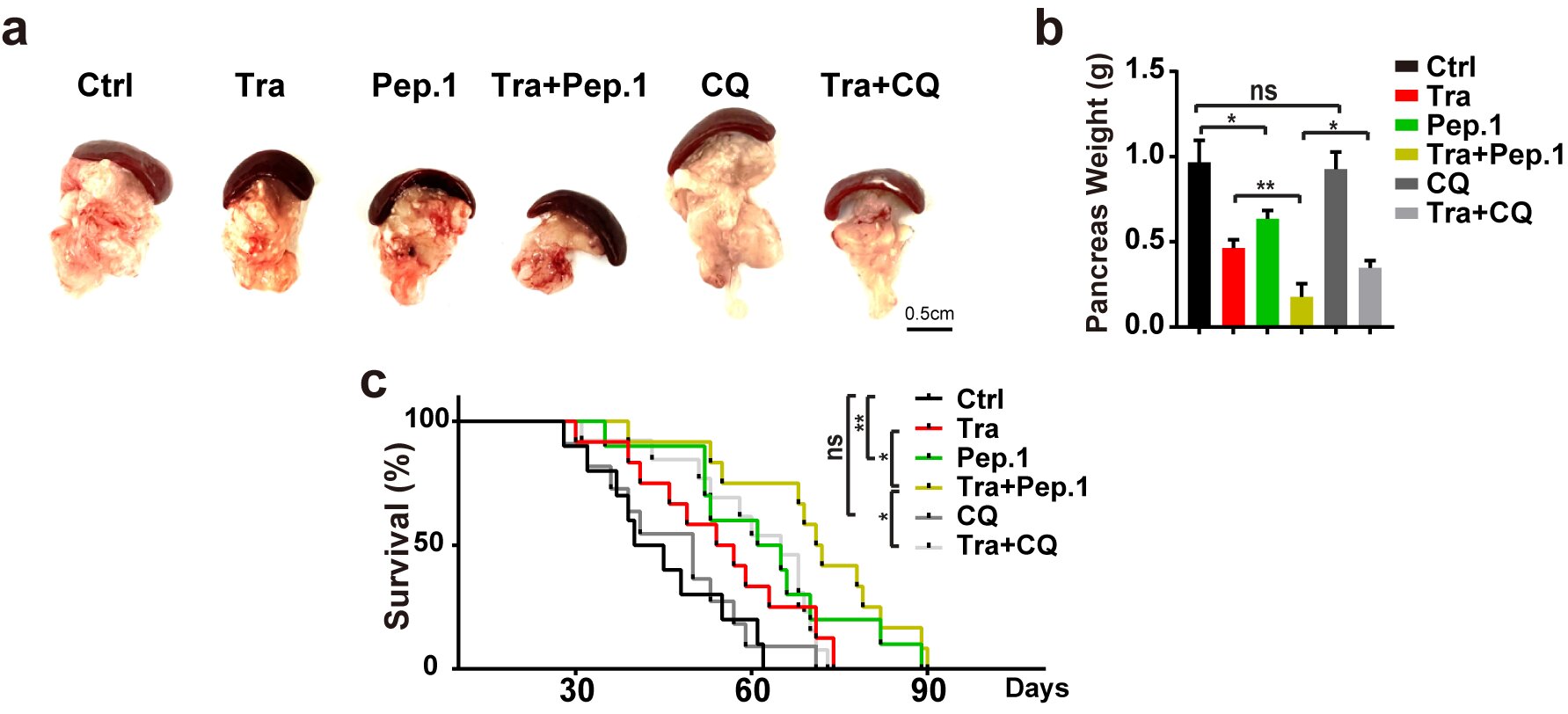
图3.Tat-Pep1抑制KPC小鼠肿瘤生长并延长其生存期
清华大学生命科学学院副教授葛亮、药学院副教授张敏和广州医科大学教授冯杜为论文通讯作者。清华大学生命科学学院博士后王晓娟、博士后李树林和广州医科大学2021级博士生林士茵(已毕业)为论文第一作者。清华大学生命科学学院教授邓海腾为该研究提供了宝贵意见,清华大学生命科学学院蛋白质中心冷冻电镜平台工程师李英、博士后韩亚平、2018级博士生王娟娟(已毕业)、2021级博士生黄智颖以及2019级本科生詹童(已毕业)为研究提供了诸多帮助。研究得到国家重点研发计划、国家自然科学基金、新基石科学基金、中国博士后科学基金、清华大学笃实基金和清华大学水木学者项目的支持。
论文链接:
https://www.nature.com/articles/s41422-025-01085-9
供稿:生命学院
编辑:李华山
审核:郭玲
© 版权声明
本文由分享者转载或发布,内容仅供学习和交流,版权归原文作者所有。如有侵权,请留言联系更正或删除。
相关文章


暂无评论...