





12345
(25S)-Δ7-Dafachronic acid(1)是一种首次在线虫中发现的类固醇激素,作为核激素受体DAF-12的配体,其在线虫生命周期和寿命调控中发挥关键作用。鉴于DAF-12信号通路在多种寄生线虫中普遍存在,(25S)-Δ7-Dafachronic acid及其衍生物展现出作为抗线虫剂的广泛应用前景。Demissidine(2)是一种从马铃薯中分离的甾体生物碱,研究表明其具有抑制肝肿瘤细胞(HepG2)生长的生物活性。此外,从天门冬中提取的甾体皂苷元Smilagenin(3)因其对阿尔茨海默病和帕金森病表现出神经保护作用而备受关注,被认为是一种具有开发潜力的苗头分子 (图1, a)。
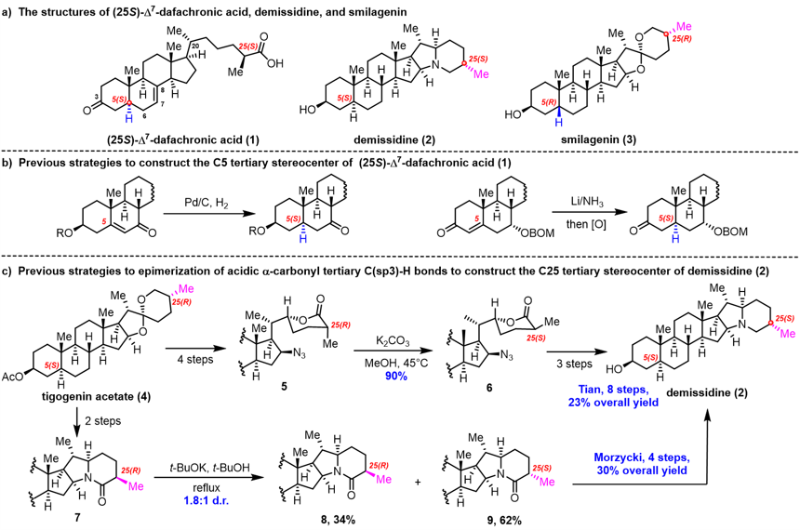
图一、(25S)-Δ7-dafachronica acid (1)、desmissidine (2)、smilagenin (3)的结构,以及目前发展的合成策略
这些分子因其独特的结构和显著的生物活性,吸引了众多合成化学家的兴趣。合成过程中,C5((25S)-Δ7-Dafachronic acid)、C25(Demissidine&Smilagenin)立体中心的构筑尤为重要,对于这一关键步骤,传统合成方法步骤繁琐,区域选择性和立体选择性难以精准控制 (图1, b-c)。因此,开发一种高效、精准的立体化学编辑策略,对于合成具有复杂结构和特定生物活性的甾体分子至关重要。近日,上海交通大学吴晶晶课题组与张兆国课题组利用光催化立体化学编辑策略,通过对甾体分子中三级C-H键的精准调控,成功实现了(25S)-Δ7-Dafachronic acid、Demissidine 和 Smilagenin的简明合成,相关成果发表在Angew. Chem. Int. Ed.上。
该策略的主要内容为通过在化合物10的C5位点进行区域选择性氢原子攫取(HAA)从而生成自由基中间体11-cis,该中间体可快速异构化为热力学更稳定的中间体11-trans。随后,氢原子供给(HAD)将沿轴向非对映选择性进行,最终实现C5位的差向异构化(图 2, a)。该策略同样适用于化合物13的C25-甲基差向异构化,使其从直立键位置异构化为平伏键位置。以(25S)-Δ7-Dafachronic acid为例,其逆合成分析如Scheme 2, c所示目标产物的C25手性甲基可通过化合物24的后期不对称氢化构建,而24可通过化合物25经光诱导脱羧烯丙基化和C7-OH消除获得,中间体25可由鹅去氧胆酸(26)通过光催化立体化学编辑、C3-OH氧化和C24羧酸缩合反应制备(图2, c)。
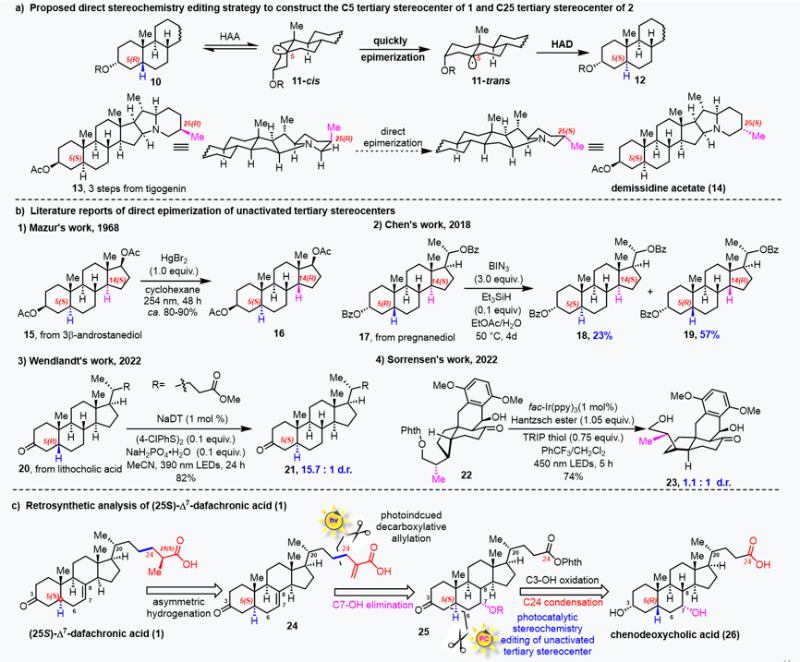
图二、光催化立体化学编辑策略以及(25S)-Δ7-Dafachronic acid分子的逆合成分析
作者首先研究了C5叔碳立体中心的直接自由基差向异构化反应。最初,作者采用了MacMillan课题组报道的β-烯胺基自由基形成策略,但未得到C5差向异构化产物。作者还尝试了氯原子和四丁基铵十钨酸盐(TBADT)等氢原子攫取试剂,但均未获得理想结果(详见SI)。受Wendlandt工作的启发,作者采用十钨酸钠(NaDT)作为氢原子攫取试剂,在真实底物上进行C5异构化实验。他们以廉价的鹅去氧胆酸(26)为起始原料,经C24酯化后,使用TEMPO/NaClO选择性氧化空间位阻较小的C3位羟基,以85%的收率得到27a。然而,在Wendlandt的标准条件下,底物27a未发生差向异构化。作者推测这是由于底物中C7-OH的存在,其C7-H键的键解离能(BDE;约92 kcal/mol)远低于C5位,导致氢原子转移更易发生在C7位。为解决这一问题,作者采用吸电子基团保护C7-OH。最初选择Ms-作为保护基,但转化率仅为15%,这可能是由于空间位阻所致。令人欣喜的是,当使用Ac-作为保护基时,底物27c在24小时内以86%的转化率得到28c。进一步添加1 mol% NaDT并延长反应时间24小时后,差向异构化比例显著提高,以95%的收率和96:4的非对映选择性得到产物28c。
在成功建立实现C5差向异构化后,作者着手合成(25S)-Δ7-Dafachronic acid(1)。通过水解和缩合两步反应,28c顺利转化为氧化还原活性酯25。随后,在作者先前报道的标准条件下,通过光诱导脱羧烯丙基化反应以71%的收率得到化合物29。使用Burgess试剂进行消除反应生成△7(8)烯烃,经水解得到丙烯酸24。最后,以张万斌教授发展的(S,Sp)-RuPhox-Ru为手性催化剂、NaHCO3和PPh3存在下,于20 bar H2压力下对丙烯酸进行不对称氢化,成功构建了C25手性甲基,反应具有优异的收率和非对映选择性(d.r.>20:1)。由此,作者以总步骤10步、40%的总收率实现了(25S)-Δ7-Dafachronic acid(1)的简洁合成(图3)。
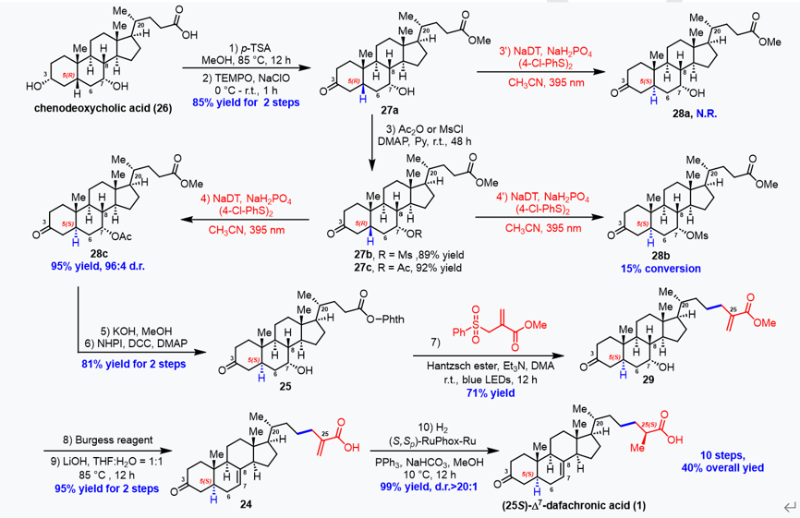
图三、从鹅去氧胆酸合成(25S)-Δ7-dafachronic acid的十步法合成路线
在实现了(25S)-Δ7-Dafachronic acid(1)的高效合成后,作者进一步将立体化学编辑策略应用于Demissidine(2)和Smilagenin(3)的合成中。由于α-氨基位置的C-H键BDE低于C25-H,因此要想实现C25-H的选择性氢原子攫取面临巨大挑战。对于这一难题,作者设想使用质子酸将氨基质子化,使α-氨基氢发生失活,从而实现C25-H的选择性氢原子攫取。作者首先采用Morzycki报道的三步法从tigogenin醋酸酯(4)合成13,正如作者预测,化合物13在标准条件下直接进差向异构化反应未能成功。在使用硫酸质子化后,差向异构化反应顺利进行,以93%的收率和大于20:1的非对映选择性得到C25差向异构化产物14。最后14再经过一步水解,这样作者就以总步骤5步、48%的总收率高效获得目标天然产物demissidine(图4)。
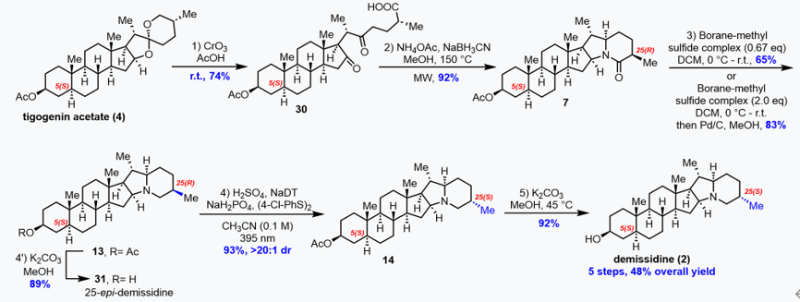
图四、从tigogenin醋酸酯合成地demissidine的五步合成路线
至此,作者成功应用立体化学编辑策略将A/B环顺式稠合的底物27c转化为热力学更稳定的A/B环反式稠合产物28c,并将13的C25-甲基由轴向位置变化到能量更有利的平伏位置。随后,作者研究了该策略在同时具有A/B环顺式稠合体系和C25-甲基直立位置的同一分子中是否表现出区域选择性。作者认为,NaDT作为一种大位阻的氢原子攫取试剂,倾向于从位阻较小的位置攫取氢原子。因此,反应预计首先发生在C25位,随后在位阻较大的C5位发生差向异构化。为验证这一假设,作者使用sarsasapogenin醋酸酯32进行光化学立体化学编辑反应。正如预期,C25差向异构化产物smilagenin醋酸酯(33)以88%的收率获得,而同时发生C25和C5差向异构化的产物tigogenin醋酸酯(4)仅得到2%的收率。化合物33经水解后,以高产率得到smilagenin(3)(图5)。
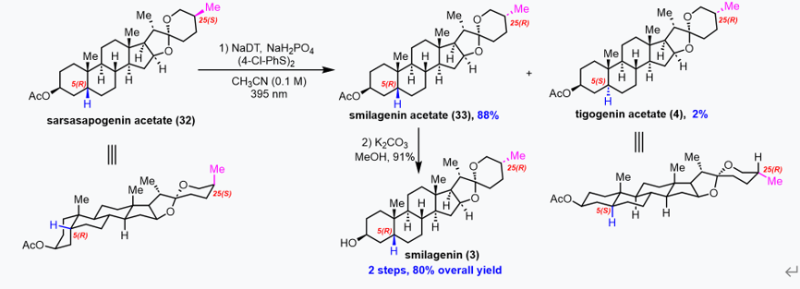
图五、smilagenin的简明合成
综上所述,通过对甾体化合物光化学立体化学编辑策略的研究,作者实现了(25S)-Δ7-dafachronic acid、demissidine和smilagenin的简洁合成。这是迄今为止这些天然产物最高效的合成路线,展示了立体化学编辑在提高天然产物合成效率方面的显著影响。通过在(25S)-Δ7-Dafachronic acid合成中采用乙酸酯保护羟基,以及在demissidine合成中对氨基进行质子化,为有效解决氢原子攫取的区域选择性问题提供了可靠的解决方案。此外,通过对sarsasapogenin醋酸酯立体化学编辑研究,作者证明了可以利用空间位阻效应调控该策略的区域选择性。
该研究成果近日以“Photocatalytic Stereochemical Editing for the Concise Syntheses of (25S)-Δ7-Dafachronic acid, Demissidine and Smilagenin”为题,在国际著名期刊Angew. Chem. Int. Ed.上线发表(DOI: 10.1002/anie.202500341)。上海交通大学博士研究生李晓童为论文第一作者,指导老师为上海交通大学吴晶晶副教授和张兆国教授。作者特别感谢上海有机所的田伟生研究员、桂敬汉研究员,上海交通大学的张万斌教授,以及新加坡南洋理工大学的Philip. S. Grant教授所提供的帮助。
原文链接:https://doi.org/10.1002/ange.202500341
作者: 吴晶晶课题组 供稿单位: 变革性分子前沿科学中心
© 版权声明
本文由分享者转载或发布,内容仅供学习和交流,版权归原文作者所有。如有侵权,请留言联系更正或删除。




1234
相关文章




12

暂无评论...